

CONCEITO:
As glicogenoses, também chamadas
dextroses ou doenças do armazenamento de glicogênio, são enfermidades
secundárias a um erro no metabolismo, o qual resulta em concentrações alteradas
de glicogênio no organismo, principalmente no fígado e nos músculos.
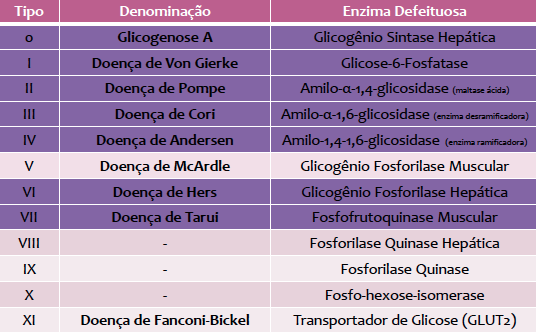
Existem mais de 12 diferentes
tipos, dependendo do defeito enzimático encontrado levando em consideração os
principais tecidos afetados. A condição foi primeiramente constatada por Von
Gierke em 1929 e, por isso, uma das formas dessa deficiência foi denominada de
Doença de Von Gierke.
CLASSIFICAÇÃO
ETIOLOGIA:
As glicogenoses podem ser
genéticas (herança autossômica recessiva) ou adquiridas.
· As de causas genéticas devem-se a um erro
congênito do metabolismo em virtude da ausência ou deficiência enzimática
envolvida na síntese ou degradação do glicogênio.
· As
adquiridas são causadas por intoxicação com o alcaloide castanospermina.
FISIOLOGIA:
O glicogênio é a forma através da
qual o organismo armazena a glicose. Ele está presente em todas as células
animais, sendo mais abundante no fígado e nos músculos. A glicose ingerida
chega ao fígado pela veia porta. Quando há necessidade de uso da glicose,
ocorre uma “quebra” do glicogênio por meio de processo enzimático ocorrendo
então liberação dela para a circulação sanguínea. Dessa forma, o fígado
proporciona a liberação de glicose para vários órgãos, incluindo o cérebro.
Quando o glicogênio não consegue
ser “quebrado” devido à deficiência de algumas das enzimas envolvidas, este se
acumula no órgão e o organismo sofre as consequências desse excesso e da falta
de glicose. Existem onze doenças distintas de armazenamento de glicogênio. A
deficiência da síntese de glicogênio, embora não resulte no armazenamento de
glicogênio, é frequentemente classificada como glicogenose tipo 0, porque pode
causar problemas semelhantes.
PRINCIPAIS SINTOMAS:
As deficiências enzimáticas
resultam em maior concentração do glicogênio em diversos tecidos do organismo.
As glicogenoses são classificadas em dez tipos diferentes, nomeadas de acordo
com o defeito enzimático específico de cada uma e os órgãos afetados.
A glicogenose tipo I representa
cerca de 25% do total das glicogenoses e é caracterizada pela deficiência da
enzima glicose-6-fosfatase, responsável pela reação final do processo de
glicogenólise e gliconeogênese. Portanto, os indivíduos com esta doença são
incapazes de converter a glicose-6-fosfato em glicose livre para a circulação
sanguínea, resultando no acúmulo excessivo de glicogênio no fígado, rins e mucosa
intestinal.
A diferenciação clínica entre as
glicogenoses às vezes é difícil, mas o tipo I é tomado aqui como exemplo por
ser o mais grave e o de início mais precoce. A hipoglicemia, convulsão,
hepatomegalia, cetose e acidemia láctica já podem estar presentes desde o
período neonatal ou aparecerem após alguns meses de vida. O fígado tem
consistência habitual, superfície lisa, mas pode atingir a fossa ilíaca direita
e esquerda.
Alguns sintomas, são: fácies de
boneca, sangramentos nasais frequentes, diarreia intermitente, retardo de
crescimento, adiposidade exagerada e musculatura diminuída. Com algum tempo de
evolução da doença, pode-se encontrar: xantomas, alteração da retina,
pancreatite, cálculo na vesícula biliar, hipertensão pulmonar devido à produção
anômala de aminas vasoativas como serotonina e doença renal crônica com
nefromegalia e hipercalciúria.
O adenoma hepático geralmente
ocorre durante ou após a puberdade, mas pode se desenvolver em qualquer idade e
se transformar em hepatocarcinoma após muitos anos.
DIAGNÓSTICO:
O diagnóstico deve partir dos
sintomas, mas como a glicogenose pode simular um quadro respiratório, a
gasometria arterial ou venosa e a ausculta pulmonar são indispensáveis. A
medida da glicemia e a constatação de hepatomegalia auxiliam no diagnóstico.
Para complementar o diagnóstico, associa-se o quadro clínico a testes de
tolerância oral à glicose e lactato, em jejum e após dieta rica em carboidrato.
O lactato é muito alto em jejum e
diminui após a administração de glicose na dieta. Esses pacientes também
apresentam triglicérides, colesterol, ureia e transaminases discretamente
elevadas no sangue. A biópsia hepática com dosagem da atividade enzimática e a
análise molecular tornam o diagnóstico definitivo.
TRATAMENTO:
Para se manterem equilibradas, as
crianças com glicogenose dependem de fonte exógena de glicose. Por isso, quando
submetidos a um jejum prolongado, como no caso de procedimentos cirúrgicos, por
exemplo, devem receber infusão endovenosa de glicose. A terapia nutricional deve
ser constituída por alimentação sem açúcares de rápida absorção e com produtos
ricos em amido. A utilização de infusão intragástrica contínua durante a noite
geralmente é necessária na fase inicial até a adaptação à dieta.
A ingestão de alimentos deve ser
fracionada em 5 a 6 refeições durante o dia. Alguns autores aconselham que o
aleitamento materno seja feito a cada 3 horas, junto com a suplementação de
glicose diluída em água. A introdução de outros alimentos pode ter início entre
4 e 6 meses de idade, devendo ser dada prioridade aos alimentos ricos em amido.
A dieta com alto teor de proteína não é aconselhada.
Alimentos e medicações com
galactose e frutose devem ser evitados e utilizadas fórmulas alimentares
infantis sem lactose. A suplementação de vitaminas e sais minerais,
principalmente de cálcio, pode ser necessária devido à limitada ingestão de
leite e frutas. A hiperuricemia necessita de medicações específicas.
Um transplante renal pode ser
necessário, no caso de evolução para a insuficiência renal crônica. O
transplante hepático é recomendado para crianças com importante déficit de
estatura, as que não responderam ao tratamento dietético ou quando existe
adenoma hepático, devido à possibilidade da transformação maligna.
REFERÊNCIAS BIBLIOGRÁFICAS
FERREIRA, Cristina T.; CARVALHO, Elisa de; SILVA, Luciana R. Glicogenoses. In: FAGUNDES, Eleonora D. T.; FERREIRA, Alexandre R.; ROQUETE, Mariza L. V. Gastroenterologia e hepatologia em pediatria: diagnóstico e tratamento. Rio de Janeiro: MEDSI EDITORA MÉDICA E CIENTÍFICA LTDA, 2003. cap. 49, p. 645-657.
FERREIRA, Cristina T.; CARVALHO, Elisa de; SILVA, Luciana R. Outras doenças metabólicas do fígado. In: BEZERRA, Jorge A. Gastroenterologia e hepatologia em pediatria: diagnóstico e tratamento. Rio de Janeiro: MEDSI EDITORA MÉDICA E CIENTÍFICA LTDA, 2003. cap. 50, p. 660-663.
SCRIVER, Charles R. et al. Glycogen Storage Diseases In: CHEN, Yuan-Tsong; BURCHELL, Ann; The metabolic and molecular bases of inherited disease. New York: THE MCGRAW-HILL COMPANIES, INC, 1995. cap. 24, p. 935-965. 7ed. Volume 1.
BRAUNWALD, Eugene et al. Doenças de depósito de glicogênio e outros distúrbios hereditários do metabolismo dos carboidratos. In: CHEN, Yuan-Tsong. Harrison: medicina interna. Rio de Janeiro: MCGRAW-HILL INTERAMERICANA DO BRASIL LTDA, 2001. cap.350, p. 2426-2433. 15 ed. Volume 2.
Nenhum comentário:
Postar um comentário